New Resources for Tuberculosis
Kim Andrews, James Hadfield, Victor Lin, Jover Lee, Jennifer Chang
Nextstrain has now released continually updated genomic surveillance resources for Mycobacterium tuberculosis, the bacterium that causes tuberculosis (TB). Results of these resources are available at nextstrain.org/tb/global (Fig. 1).
TB is a major global health issue, causing more deaths around the world than any other infectious disease (WHO Global tuberculosis report 2024). About a quarter of the world's population is estimated to have been infected with M. tuberculosis, although in most cases the infection is latent, with only 5-10% of infected individuals expected to fall ill. TB is curable through antibiotic treatment, but the emergence of drug resistant strains of M. tuberculosis has complicated our ability to control this disease on a global scale.
Our new Nextstrain M. tuberculosis genomic surveillance resources aim to contribute to global monitoring of this pathogen by providing continually updated phylogenetic analyses using publicly available sequence data for strains from across the world. Using community-provided tools, we also predict whether strains are drug resistant and classify strains by phylogenetic lineage (Fig. 1). The analysis is updated every week using a random subset of approximately 1000 M. tuberculosis samples from across the world over time from the NCBI SRA. The phylogeny includes a random subsample from all lineages in the M. tuberculosis complex that have available sequence data, including both human-adapted and animal-adapted lineages. Mycobacterium canettii is excluded from the phylogeny due to its high genetic divergence from other lineages.
A. Drug resistance types
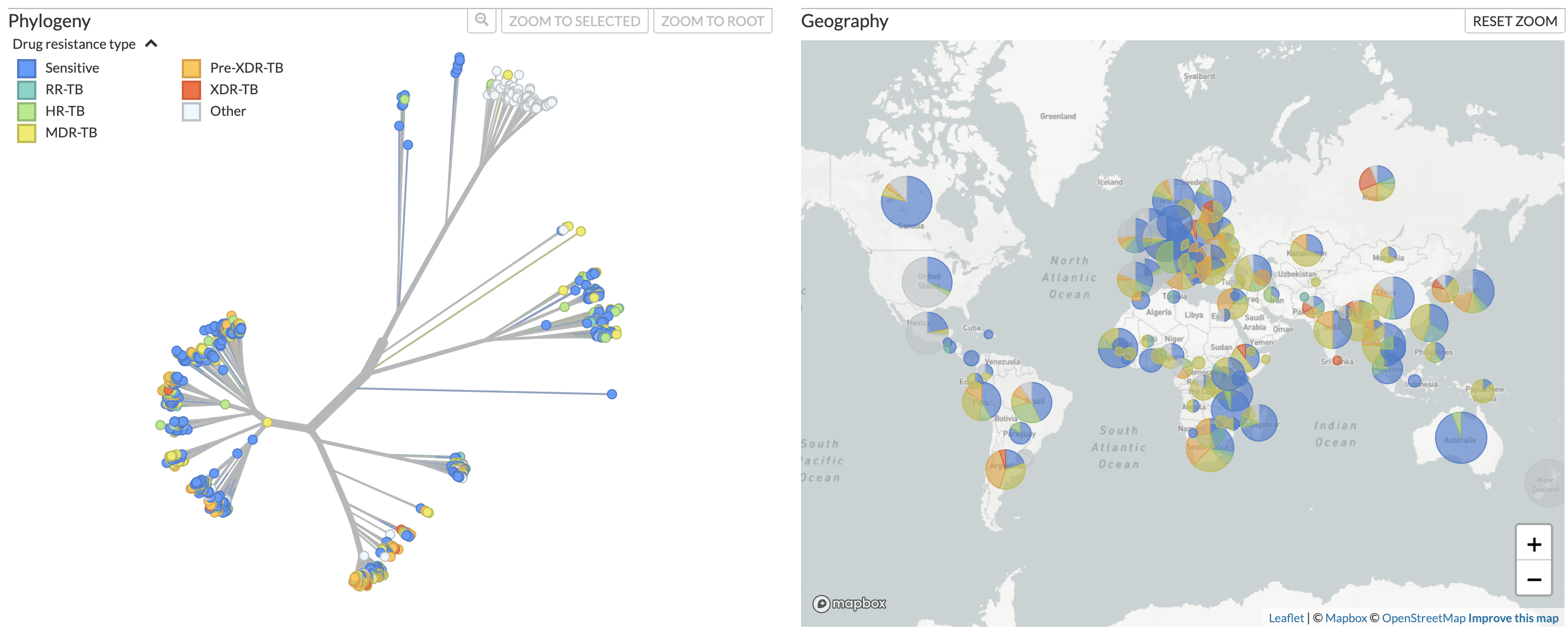 B. Lineage assignments
B. Lineage assignments
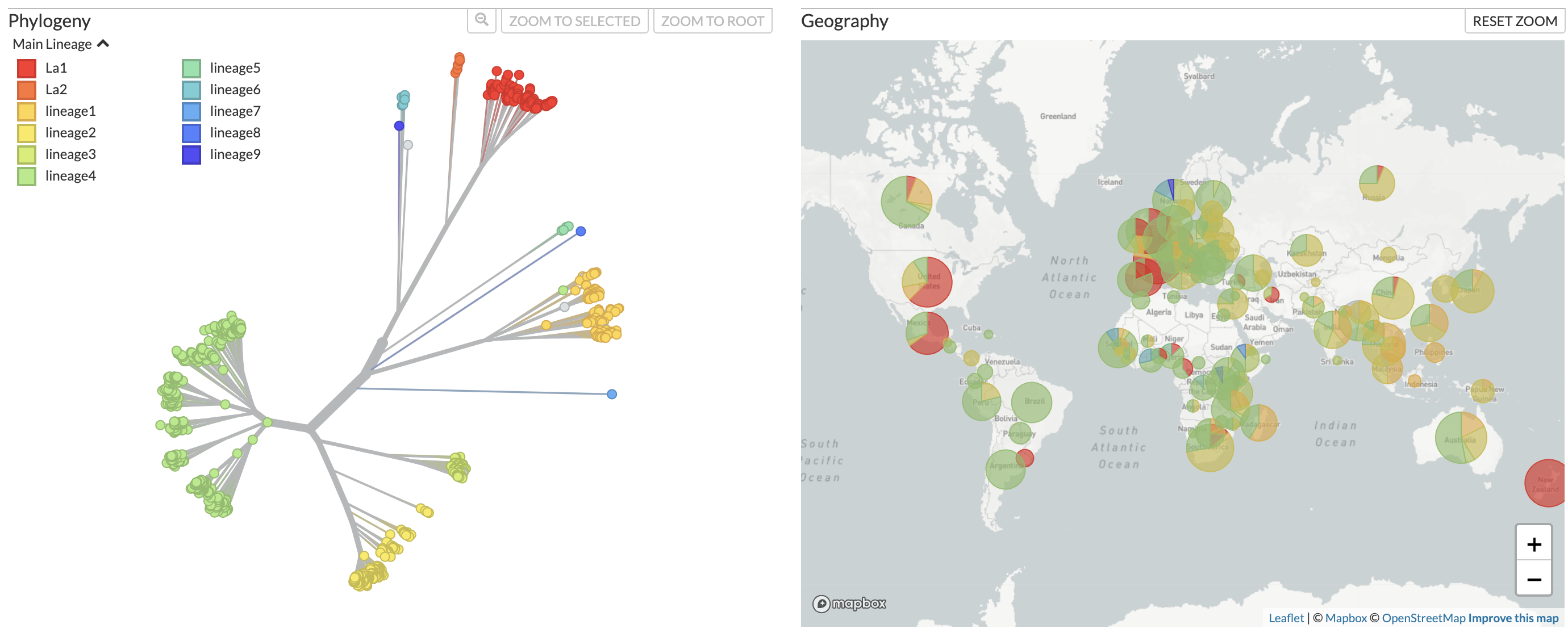 Figure 1. Phylogenetic tree of Mycobacterium tuberculosis samples from across the world alongside a map showing distribution of the samples, available at nextstrain.org/tb/global. Samples can be colored by A) predicted drug resistance and B) Lineage assignments.
Figure 1. Phylogenetic tree of Mycobacterium tuberculosis samples from across the world alongside a map showing distribution of the samples, available at nextstrain.org/tb/global. Samples can be colored by A) predicted drug resistance and B) Lineage assignments.
# Nextstrain's first bacterial real-time analysis
These M. tuberculosis genomic surveillance resources represent Nextstrain's first real-time analyses for a bacterial pathogen. Although these resources share the same outputs as our viral pathogen resources, the underlying analyses differ in many ways to accommodate the distinct genomic characteristics of bacterial pathogens in general, and M. tuberculosis in particular.
One of the main differences is that the workflow starts from raw Illumina sequence reads for each sample, whereas our viral workflows start from genome assemblies (Fig. 2). The raw sequence reads are aligned to a reference genome and are then used to identify variable sites within each genome using the program snippy. Variable sites are then summarized in a VCF file that is used as input for the phylogenetic analysis. This is in contrast to our viral workflows, for which the phylogenetic input is a full genome alignment in a FASTA file.
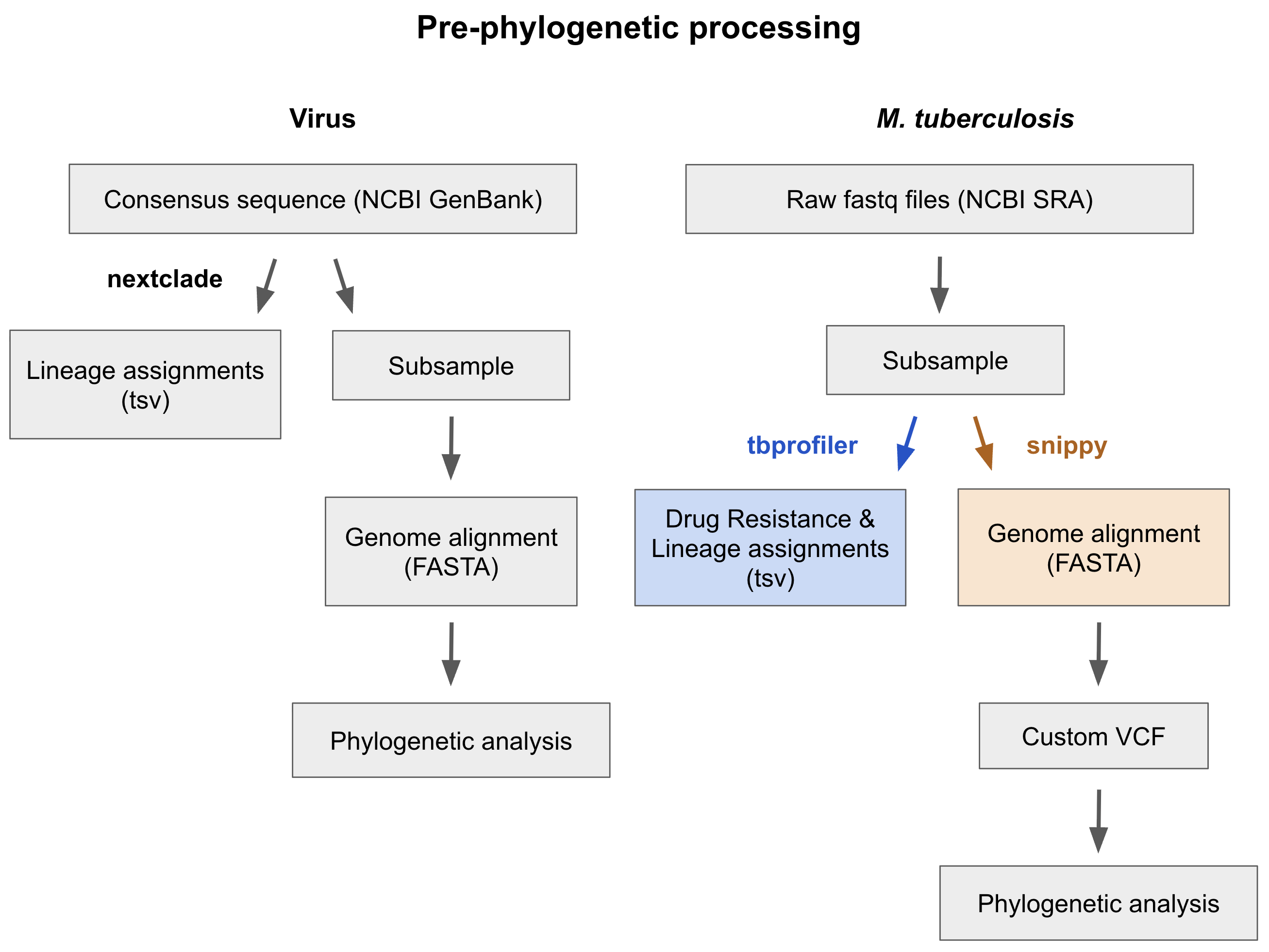 Figure 2. Comparison of analysis steps in Nextstrain real-time genomic surveillance resources for a typical viral workflow versus the Mycobacterium tuberculosis workflow, focusing on steps conducted prior to phylogenetic analysis.
Figure 2. Comparison of analysis steps in Nextstrain real-time genomic surveillance resources for a typical viral workflow versus the Mycobacterium tuberculosis workflow, focusing on steps conducted prior to phylogenetic analysis.
Our M. tuberculosis workflow also uses the program TBProfiler (Phelan et al. 2019, Verboven et al. 2022) to predict resistance to anti-tuberculosis drugs by comparing the genome sequences of each sample against the tbdb reference database, which has a list of mutations associated with drug resistance published by the World Health Organization and other sources (Fig. 2). TBProfiler also assigns a phylogenetic lineage to each sample using a reference database of lineage-specific mutations. Drug resistance and lineage classifications are provided as options for coloring on the phylogeny.
The M. tuberculosis workflow requires substantially more computational resources than most of our viral workflows due to 1) the much larger genome size of bacteria compared to viruses, and 2) the use of raw sequence read files, which are much larger than genome assembly files. One way we address these computational requirements is by using Amazon Web Service (AWS) high-performance computing resources for our weekly runs. Every time we run the analysis, TBprofiler and snippy results are generated and stored on AWS for each of the samples that was randomly selected for the analysis. The next time the analysis runs with a new random subsample, the TBprofiler and snippy results for any sample that was previously analyzed are downloaded without having to re-run TBprofiler and snippy.
# Acknowledgments & request for comments
We gratefully acknowledge the authors, originating and submitting laboratories of the genetic sequences and metadata for sharing their work. Additionally, we welcome comments or suggestions from TB researchers on how to improve these Nextstrain datasets for their use case.










