New Resources for Rubella
John SJ Anderson
We are pleased to announce the availability of a Rubella phylogenetic analysis and dataset on Nextstrain.org, as well as a Rubella Nextclade dataset. We provide phylogenetic trees based on both whole genome analysis, as well as analysis of the E1 region used in the WHO genotype reference. These trees are automatically rebuilt with new Rubella sequences as they are deposited in NCBI. We welcome feedback from Rubella researchers about any aspect of either the Nextclade dataset or the phylogenetic trees on Nextstrain.org.
# Phylogenetic analysis
Rubella virus (Rubivirus rubellae) is a positive-strand RNA virus with a 9,762 base genome. It causes rubella, a formerly common respiratorily-transmitted illness, primarily infecting children. When rubella infects a pregnant person and spreads to the fetus, it causes congenital rubella syndrome (CRS), which can lead to a wide variety of congenital defects. The earlier in pregnancy infection happens, the more severe these defects tend to be. Rubella has been effectively eradicated in many parts of the world due to successful vaccination programs. Available vaccines provide lifelong immunity after a single dose, usually administered as part of the tri-valent "MMR" vaccine, which also protects against measles and mumps.
The Rubella genome has two open reading frames (NSP and SP), the latter of which encodes three structural proteins, C (capsid), E1, and E2 (envelope proteins). E1 is the source of hemagglutin activity for the virus, and is the only region that is sequenced for many samples. It is also the basis of the WHO genotype reference, which we used for genotype definition in the Nextclade dataset. The WHO nomenclature classifies Rubella into have two large overarching clades (1 and 2), split into specific genotypes (1A-1J and 2A-2C) based on clades observed in trees built with reference samples. (See WHO genotype reference for further specifics.)
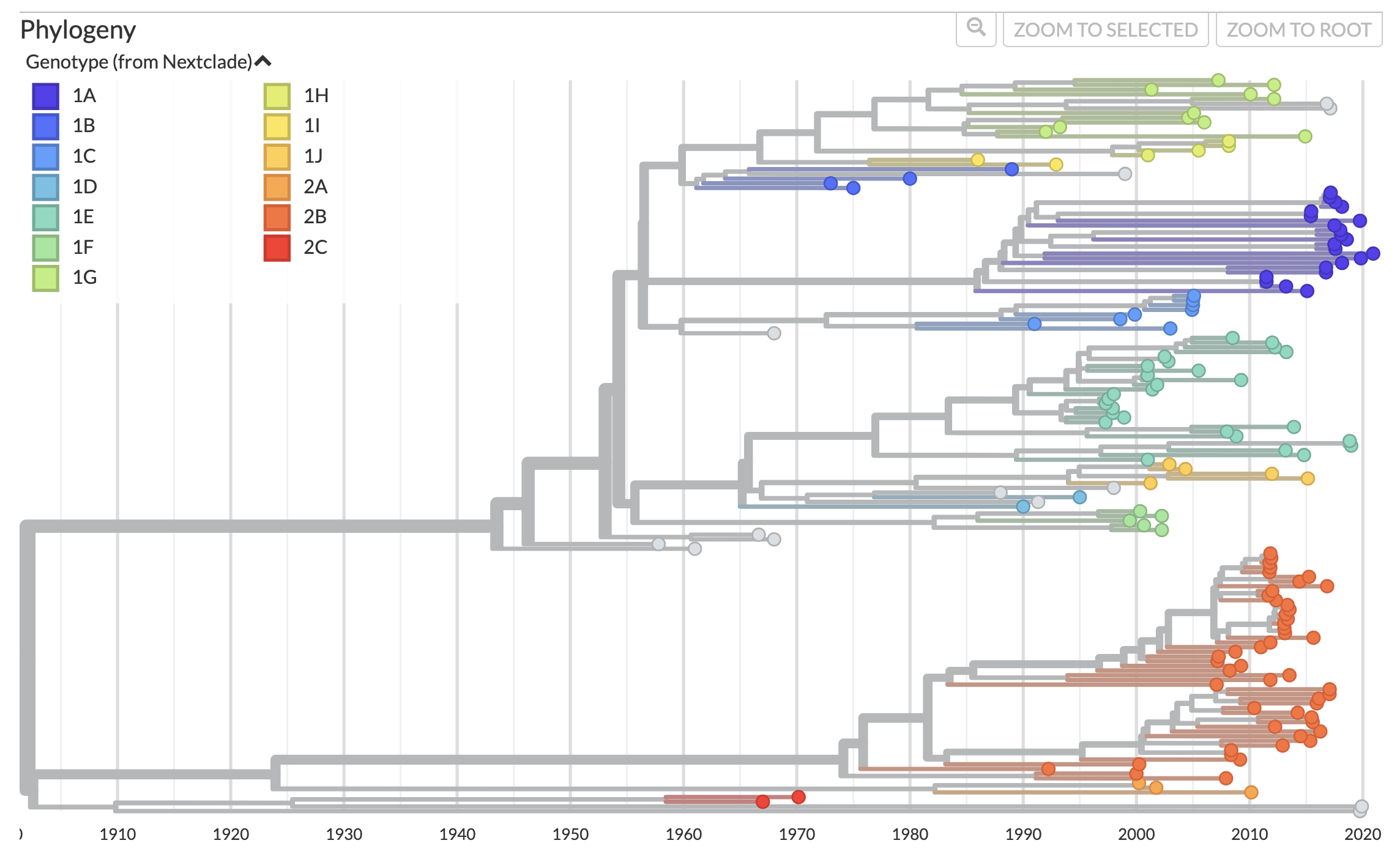 Figure 1. Phylogenetic tree of rubella virus whole genome. The
phylogenetic tree is colored by genotype, based on reference strains
from the WHO genotype reference. Available at
nextstrain.org/rubella.
Figure 1. Phylogenetic tree of rubella virus whole genome. The
phylogenetic tree is colored by genotype, based on reference strains
from the WHO genotype reference. Available at
nextstrain.org/rubella.
The Nextstrain phylogenies support coloring by both genotypes assigned via the Nextclade dataset, as well as genotypes extracted from parsing strain names and other metadata present in GenBank records. These two methods are broadly, but not 100%, congruent with each other. We also have included a coloring for the specific WHO reference strains, to facilitate visualizing how these strains are positioned in the broader genomic and E1 gene trees.
# Nextclade dataset
We developed an initial version of the Nextclade dataset based strictly on the 32 reference samples from the WHO genotype reference. This initial dataset did not do an adequate job of assigning genotypes to the full trees, so we revised it with the addition of 32 other samples. The genotypes for these samples were based on both annotations in GenBank, and their position in the phylogenetic trees relative to the WHO strains. In general, we tried to select additional samples that were more basal to the WHO samples in the tree, with the intention of capturing additional genotype diversity.
The 1A genotype, as noted in the original WHO nomenclature publication, is polyphyletic. In particular, we observed a clade of approximately 22 samples in the whole genome tree, which appear to reflect more recent vaccine-derived 1A strains, which have a more distal placement in the tree relative to most of the original 1A reference strains. We have elected to make the Nextclade dataset's definition of the 1A genotype based on this more derived clade, rather than the more basal original samples, which do not group with any more recent samples in either the whole genome or E1 region specific trees.
# Nextstrain resources
We curate sequence data and metadata from NCBI as the starting point for our analyses. Curated sequences and metadata are available as flat files at:
- data.nextstrain.org/files/workflows/rubella/metadata.tsv.zst
- data.nextstrain.org/files/workflows/rubella/sequences.tsv.zst
# Acknowledgments & request for comments
We gratefully acknowledge the authors, originating and submitting laboratories of the genetic sequences and metadata for sharing their work. Additionally, we welcome comments or suggestions from rubella researchers on how to improve these Nextstrain datasets for their use case.










