Nextstrain Annual Update March 2025
Trevor Bedford, Richard Neher
We continue to follow a model of genomic surveillance in which our central conceit has two pillars:
Routine real-time genomic surveillance across a variety of circulating pathogens
Rapid pivots to emerging public health threats
as we believe that proper tooling for real-time surveillance (1) enables rapid response when new threats emerge (2).
# Recent activities (March 2024 to March 2025)
# Pathogen-specific analyses
We've continued our tracking of evolution of SARS-CoV-2 at /ncov, with nine new clades through 2024 and into 2025. We've continued to estimate multinomial logistic regression (MLR) fitness models and display these inferences for clades and lineages at /sars-cov-2/forecasts. We display these lineage fitnesses on the tree via a new "MLR lineage fitness" coloring. We also revamped default clade coloring to better distinguish between circulating clades. Tracking of seasonal influenza at /seasonal-flu proceeds in a similar fashion, with emphasis on keeping clade nomenclature updated.
In response to the H5N1 cattle outbreak, we set up an automated analysis that sources data from SRA and GenBank. The USDA does not immediately upload complete genomes with US state and collection date metadata and instead uploads unannotated raw reads to SRA. The Andersen Lab has been assembling these reads into consensus genomes. We ingest and deduplicate across these data sources. More recent zoonoses of genotype D1.1 into cattle have also been automated and shared in a similar fashion. In addition, Moncla Lab has continued to keep pace with H5N1 evolution in birds, including a narrative showcasing the evolution and spread of H5N1 in North America.
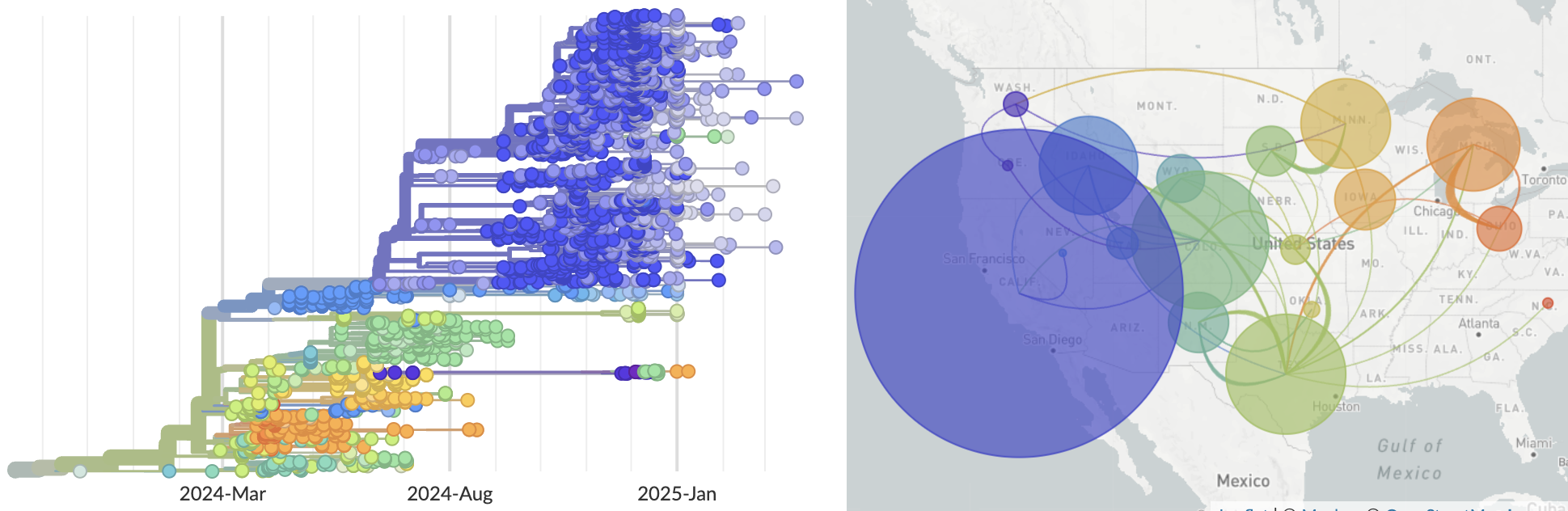
This past year, we devoted a fair amount of effort to constructing baseline analyses across endemic and emerging viral pathogens. For each pathogen we provide a GitHub repository that contains workflows for automated data ingest from NCBI GenBank, a pipeline to generate Nextclade datasets, and automated phylogenetic analysis. This year we completed automated builds for
- Dengue virus at /dengue
- Lassa virus at /lassa
- Metapneumovirus at /hmpv
- Measles virus at /measles
- Oropouche virus at /oropouche
- Rabies virus at /rabies
- Seasonal coronaviruses at /seasonal-cov
- West Nile virus at /WNV
- Yellow Fever virus at /yellow-fever

The INRB of the Democratic Republic of the Congo shared an mpox clade I analysis using their genomic surveillance data.
# Improvements to bioinformatic tooling
Improvements to Nextclade included the ability to view mutations and substitutions of query sequences relative to the reference sequence, clade founder sequences, and lineage founder sequences. Furthermore, individual strains, for example vaccine antigens, can be designated alternative references to compare to. One can now also use arbitrary Nextstrain trees as Nextclade datasets. Several Nextclade datasets have been contributed by the community, including datasets that allow assignment of the recently proposed consensus nomenclature for dengue, the IAV H5N1 nomenclature, and PRRSV-1 and 2.
In Augur, we've added the additional command augur merge to combine across rows and columns in multiple metadata sources, provided the ability to perform weighted subsampling in augur filter, included additional augur curate subcommands, implemented Excel support for input to augur curate, added a --remove outgroup flag to augur refine, added helpful commands for augur read-file and augur write-file and include a debug mode via AUGUR_DEBUG environment variable.
In Auspice, we've added a focus on selected toggle to highlight selected tips on the tree and linkouts to other platforms including Taxonium, MicrobeTrace and for ad hoc Nextclade datasets. Additionally, we've made improvements to display of measurements data, to display of tangletrees, to display of trait confidence values and to general Auspice performance. Finally, we've done considerable work to implement a big new visualization feature we're calling "streamtrees" that allows collapsing of clades / lineages into streams highlighting samples through time and sample metadata. This is designed to improve scaling and visual legibility when there are many thousands of samples in an analysis. This is not yet shipped, but we hope it will be soon.
# Other work to highlight
With nextstrain.org, we've sought to surface timely and relevant analyses and also to improve UI behind dataset search. This has manifested as a revamp to the splash page particularly to include a set of "showcase tiles" highlighting recent and relevant analysis from the Nextstrain team and external groups. We've provided similar tiles for all core pathogens listed at /pathogens. Additionally, we've provided a "cards" UI for all datasets listed at /pathogens and /groups that surfaces dataset hierarchy and frequency / history of dataset specific updates.
This year, we conducted training at VEME in Brazil and presented work at Virus Genomics, Evolution and Bioinformatics in the UK.
# Planned directions for the future
In planning work for the March 2025 to March 2026 period, we've emphasized infrastructural priorities that position us in the best way to respond to emerging outbreaks, epidemics and pandemics. This includes:
Support for large datasets via streamtrees and other improvements to Auspice. As noted above, a first version of streamtrees is almost live. However, this will need further refinement in Auspice and will need work in Augur to support delineation of evolutionarily or epidemiologically relevant clades. Future pandemics or impactful epidemics will have a deluge of data and supporting trees with more than ~4000 tips is a key priority. With streamtrees in place, existing Augur workflows should be able to handle 20k-30k datasets without too much difficulty.
Support for frequencies analyses and visualization. We've continued with live SARS-CoV-2 frequencies fitness analysis and visualization at /sars-cov-2/forecasts and separately with influenza frequencies visualization at github.com/nextstrain/flu_frequencies. Frequencies analysis (as opposed to phylogenetic analysis) allows nearly arbitrary scaling to dataset size and greatly facilitates analysis of lineage and mutation fitness. We believe that a system of automated nomenclature + streamtrees view in Auspice + frequencies view would provide a foundation for large-scale evolutionary analysis. We didn't make much progress on this Aim over the previous year, but we hope to establish a clear direction here in 2025.
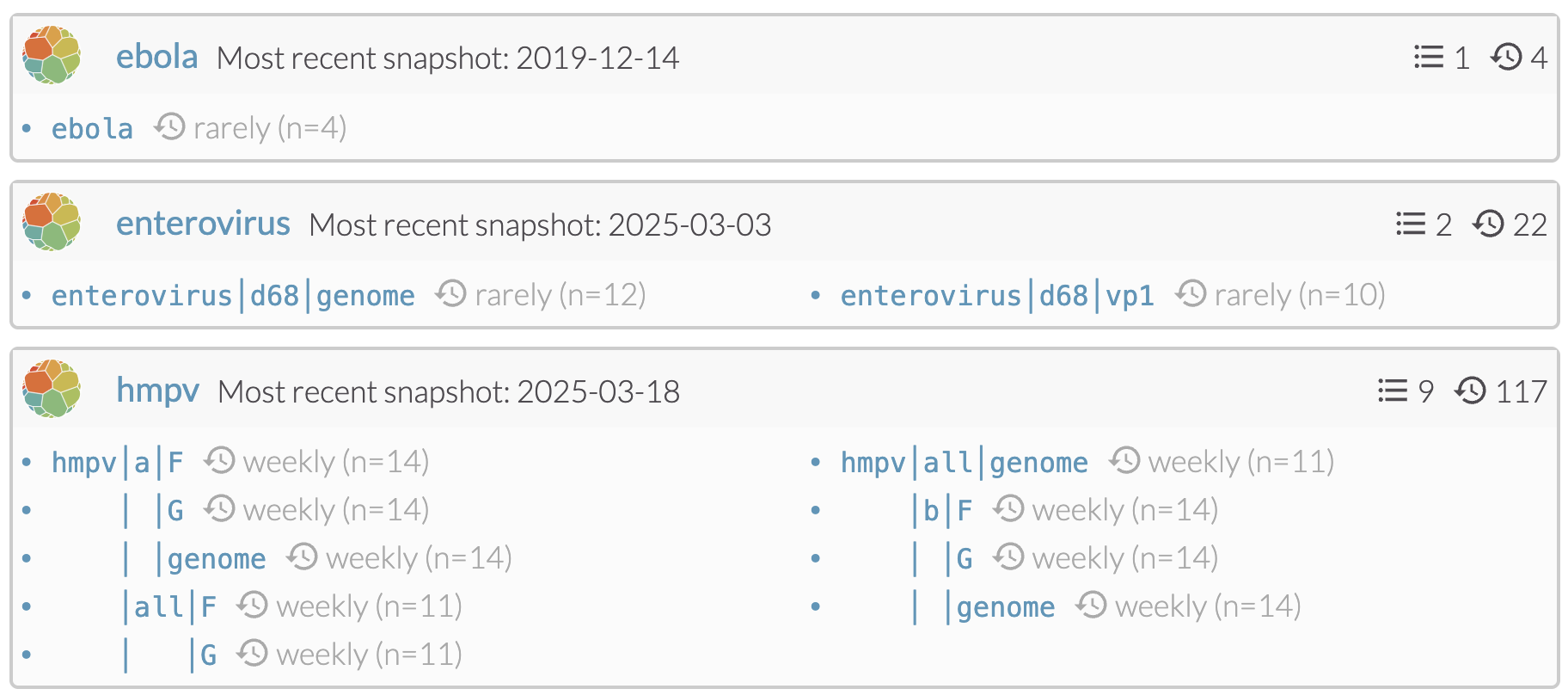
Continued build out of automated workflows to 20-30 pathogens. We believe this pattern of automated ingest via NCBI and automated phylogenetics to be a strong basis and would like to continue to establish a foundation of real-time analysis across pathogens. Currently we have 15 automated pathogens after adding an additional 9 over the course of the previous year. Current contenders for automation include CCHV, hepatitis B virus, Ebola virus, EV-D68 and tuberculosis (TB), which would bring us to 20. Work is actively proceeding on TB, which would represent the first automated bacterial workflow, though a few non-automated examples currently exist (including Y. pestis and cholera).
Improvements to Nextclade. Our primary plans for the coming year are to (i) generate annotations for query sequences that can be used to submit these sequences to NCBI, (ii) Allow users to analyze collections of sequences that map to difference reference sequences, e.g. different segments of influenza viruses, (ii) support for segmented viruses and multi-segment nomenclatures. The latter is a bigger project that we still need to scope in greater detail, but we envision such functionality to help analysis in scenarios like the recent H5N1 outbreaks.
Facilitate running pathogen workflows with user data and user config. Our current paradigm is pathogen-specific workflows (housed in individual GitHub repositories) that include ingest from NCBI. However, spiking in user data alongside NCBI data is not built into our workflows, nor is it easy to override default config. Here, we aim to provide a generic pattern for including additional user data alongside curated data as well as a generic pattern for overridable config. This will be an iterative process, with improvements that include specific Augur changes like
augur mergesequence support,augur subsample, and layering Auspice config files inaugur export. This also includes work on the Nextstrain CLI to reduce the need for users to interface with source code and Git software, which has been a continual pain point for public health professionals.Ongoing operational investment. In the mission to keep abreast of evolution across a panoply of viruses, we've come to rely on various data systems. Streamlining / improving these systems would be helpful to reduce week-to-week maintenance burden. This includes replacing the Fauna database that we still rely on for our work on seasonal influenza, speeding up
augur filter, which is a strong bottleneck on processing SARS-CoV-2 data, and revamping ncov-ingest to not re-process records each day. Additionally, although we've made good progress this year with surfacing datasets on nextstrain.org, there is additional work here to fold in narratives into the new "cards" UI, to include the cards UI across the board in places like /community and individual groups pages and finally to use a similar cards approach to surfacing not just final Auspice datasets, but also to surface intermediate data like sequence FASTAs and metadata TSVs.










