 About us
About us Core pathogens
Core pathogens SARS-CoV-2
SARS-CoV-2 Open source tooling
Open source tooling Nextstrain Groups
Nextstrain GroupsFeatured analyses



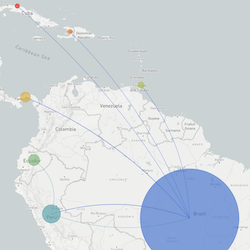
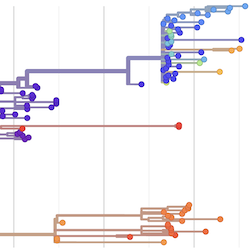
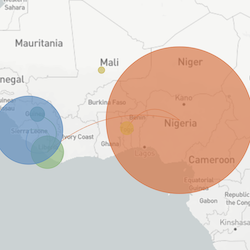
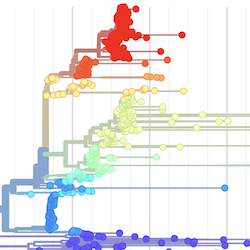

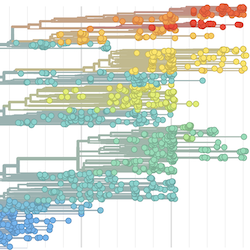
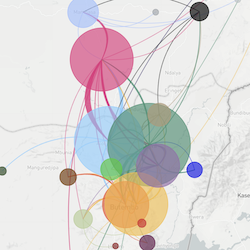

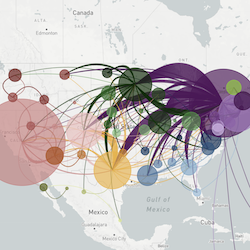
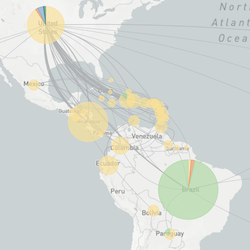
Philosophy
Pathogen Phylogenies
In the course of an infection and over an epidemic, pathogens naturally accumulate random mutations to their genomes. This is an inevitable consequence of error-prone genome replication. Since different genomes typically pick up different mutations, mutations can be used as a marker of transmission in which closely related genomes indicate closely related infections. By reconstructing a phylogeny we can learn about important epidemiological phenomena such as spatial spread, introduction timings and epidemic growth rate.
Actionable Inferences
However, if pathogen genome sequences are going to inform public health interventions, then analyses have to be rapidly conducted and results widely disseminated. Current scientific publishing practices hinder the rapid dissemination of epidemiologically relevant results. We thought an open online system that implements robust bioinformatic pipelines to synthesize data from across research groups has the best capacity to make epidemiologically actionable inferences.
This Website
This website aims to provide a real-time snapshot of evolving pathogen populations and to provide interactive data visualizations to virologists, epidemiologists, public health officials and citizen scientists. Through interactive data visualizations, we aim to allow exploration of continually up-to-date datasets, providing a novel surveillance tool to the scientific and public health communities.
Future Directions
Nextstrain is under active development and we have big plans for its future, including visualization, bioinformatics analysis and an increasing number and variety of datasets. If you have any questions or ideas, please contact us.
A bioinformatics and data viz toolkit
Nextstrain provides an open-source toolkit enabling the bioinformatics and visualization you see on this site. Tweak our analyses and create your own using the same tools we do. We aim to empower the wider genomic epidemiology and public health communities.
Support










All source code is freely available under the terms of the GNU Affero General Public License. Screenshots may be used under a CC-BY-4.0 license and attribution to nextstrain.org must be provided.
This work is made possible by the open sharing of genetic data by research groups from all over the world. We gratefully acknowledge their contributions. Special thanks to Kristian Andersen, Josh Batson, David Blazes, Jesse Bloom, Peter Bogner, Anderson Brito, Matt Cotten, Ana Crisan, Tulio de Oliveira, Gytis Dudas, Vivien Dugan, Karl Erlandson, Nuno Faria, Jennifer Gardy, Nate Grubaugh, Becky Kondor, Dylan George, Ian Goodfellow, Betz Halloran, Christian Happi, Jeff Joy, Paul Kellam, Philippe Lemey, Nick Loman, Duncan MacCannell, Erick Matsen, Sebastian Maurer-Stroh, Placide Mbala, Danny Park, Oliver Pybus, Andrew Rambaut, Colin Russell, Pardis Sabeti, Katherine Siddle, Kristof Theys, Dave Wentworth, Shirlee Wohl and Cecile Viboud for comments, suggestions and data sharing.
