Year-letter Genetic Clade Naming for SARS-CoV-2 on Nextstrain.org
Emma B Hodcroft, James Hadfield, Richard A Neher, Trevor Bedford
Nextstrain provides clade or lineage information for a number of pathogens it already tracks, including influenza and enterovirus D68. Nextstrain also introduced informal clade designations for SARS-CoV-2 on 4 March 2020, largely to aid internal discussions and to create URL links allowing ‘automatic zoom’ to an area of the tree that was of interest. These clades names were ad-hoc letter-number combinations (e.g. A2a) and were never intended to be a permanent naming system (and never visible by default). Nevertheless, these clades have been used by some to discuss different aspects of the phylogeny on Nextstrain, underscoring the need for a more long-term, formal proposal to designate SARS-CoV-2 clades.
Our objective here is to introduce a naming system that facilitates discussion of large scale diversity patterns of SARS-CoV-2 and label clades that persist for at least several months and have significant geographic spread. This objective is distinct from and complementary to Rambaut et al. 2020 (1), who proposed a dynamic system for labeling transient lineages that have local epidemiological significance. Their proposal results in a large number of short lived labels that maintain some information on the hierarchical structure and facilitate discussion of local short term dynamics, but are not intended to provide large scale structure.
Our nomenclature proposal has the following objectives:
- label genetically well defined clades that have reached significant frequency and geographic spread,
- allow for transient clade designations that are elevated to major clades if they persist and rise in frequency,
- provide memorable but informative names,
- gracefully handle clade naming in the upcoming years as SARS-CoV-2 becomes a seasonal virus.
To provide memorable and pronounceable names, we want to avoid the common pattern of stringing together alternating numbers and letters, as this pattern tends to become arcane with time, e.g. 3c2.A1b for A/H3N2 influenza. Instead, we propose to name major clades by the year they are estimated to have emerged and a letter, e.g. 19A, 19B, 20A. The yearly reset of letters will ensure that we don't progress too far into the alphabet, while the year-prefix provides immediate context on the origin of the clade that will become increasingly important going forward. We aim for ~5 clades per year. These are meant as major genetic groupings and not intended to completely resolve genetic diversity. The hierarchical structure of clades is sometimes of interest. Here, the “derivation” of a major clade can be labeled with the familiar “.” notation as in 19A.20A.20C for the major clade 20C.
Within these major clades, we will monitor potential ‘emerging clades', which we will label by their parent clade and the nucleotide mutation(s) that defines them (ex: 19A/28688C). It should be noted however, that these mutations are only meaningful in that they define the clade; no other functional or epidemiological significance should be attributed to them. Once a subclade reaches (soft) criteria on frequency, spread, and genetic distinctiveness, it will be renamed to a major clade (hypothetically 19A/28688C to 20D). The use of mutations as emergent clade labels has the desirable property that the labels stay meaningful after a renaming. Additionally, anyone can refer to any clade in the SARS-CoV-2 phylogeny using this nomenclature. For example, we can use 19A/28688C,8653T to refer to a more narrow subclade. This is obviously verbose, but is fully descriptive and has the advantage that the definition is embedded in the name itself which allows subclades to be discussed by the community without needing a centralized database to be updated.
# Definition of major clades
We propose to name a new major clade when it reaches a frequency of 20% globally. When calculating these frequencies, care has to be taken to achieve approximately even sampling of sequences in time and space since sequencing effort varies strongly between countries. A clade name consists of the year it emerged and the next available letter in the alphabet. A new clade should be at least 2 mutations away from its parent major clade.
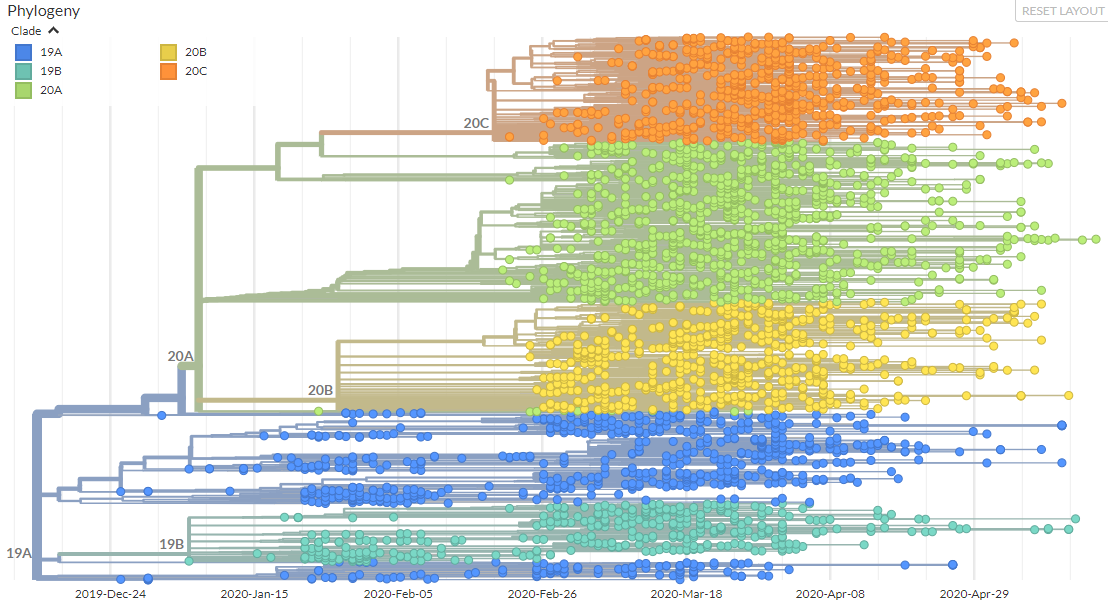 Fig 1. Nextstrain ‘global’ run with the new Nextstrain major clades labelled.
Fig 1. Nextstrain ‘global’ run with the new Nextstrain major clades labelled.
By these criteria, the first two clades are 19A and 19B which correspond to the split marked by mutations C8782T and T28144C. These clades were both prevalent in Asia during the first months of the outbreak. The next clade that was named is 20A corresponding to the clade that dominated large European outbreak in early 2020. It is distinguished from its parent 19A by the mutations C3037T, C14408T and A23403G.
After this, we've seen two further clades appear: 20B, another European clade separated clearly by three consecutive mutations: G28881A, G28882A, and G28883C and 20C, a largely North American clade, distinguished by mutations C1059T and G25563T.
The clade definitions are coded in as a tabular file that defines a genotypic signature for each clade. We provide a script that generates a table with clade assignments for a set of sequences.
Additionally, definitions of the clades, as well as a table of the current clades, with some detail on their characteristics and definition, is available on the ‘ncov’ github repository documentation (github.com/nextstrain/ncov) here. In parallel to the large-scale clade annotation, Nextstrain will support the Pangolin lineage nomenclature developed by Rambaut et al. 2020 (1), and continue to offer color-by for the ‘old’ Nextstrain clades so that exiting references to these clades are still identifiable.
# Using Nextstrain Clade Definitions
To make it easy for users to identify the Nextstrain clade of their own sequences, we provide a simple python script that can be run on any Fasta file to assign appropriate clades. This script is part of the ‘ncov’ github repository, but does not require running any other part of the pipeline. However ‘augur’ must be installed to run the script. This can be done a number of different ways, but is often most easily done using ‘pip’.
Links:
- An up-to-date description of our approach to clade names and the current clades can be found on the Nextstrain ‘ncov’ github here.
- Clades show on the current Nextstrain 'Global' SARS-CoV-2 tree can be viewed here.
- Pangolin clades can be displayed on the Nextstrain runs, as demonstrated here.
- The ‘old’ Nextstrain clades can still be viewed here as a color-by for those papers/references that have used them
References:
(1) Andrew Rambaut, Edward C. Holmes, Verity Hill, Áine O’Toole, JT McCrone, Chris Ruis, Louis du Plessis, Oliver G. Pybus. “A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology”. bioRxiv 2020.04.17.046086; doi: https://doi.org/10.1101/2020.04.17.046086










